Cynthia Ma1, Michael Brent1
1) Washington University in St Louis.
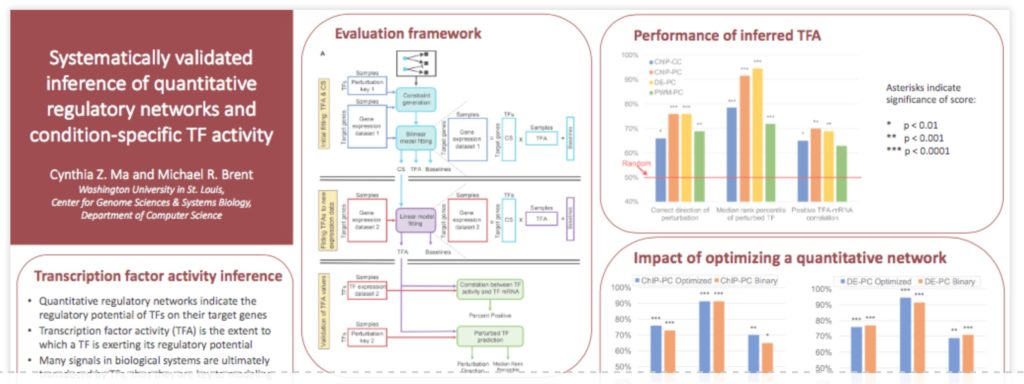
Cells process and respond to information from their environments, in part, by changing the activity levels of transcription factors – the degree to which each TF exerts its regulatory potential on its target genes. To understand the cell’s information processing, we must know which TFs change activity in response to specific extracullular stimuli or intracellular conditions. However, changes in TF activity (TFA) are difficult to measure directly because they have diverse molecular implementations, including changes in protein abundance, localization, and post-translational modifications. An alternative is to infer changes in TFA from changes in the expression levels of their target genes. This idea has been pursued in previous work, including Network Component Analysis (Liao et al 2003), RegulonProfiler (Boorsma et al 2009), and ISMARA (Balwierz et al 2014), but until now, systematic, genome-scale evaluation of such methods has not been possible. We present the first such evaluation. We find that TFA inference really does work, but only if the input network identifying the targets of each TF is of high quality. We compare multiple methods of constructing such networks, including from chromatin immunoprecipitation (ChIP) binding data, binding specificity models, or differential expression after direct perturbation of TFs. We find that perturbation response data are both necessary and sufficient for good performance, whereas the other sources are neither.
Our objective evaluation is made possible by the availability of two large data sets of gene expression after direct perturbation of TFA in yeast. One is based on knockouts of ~1,400 genes (Kemmeren et al 2014). The other is a new, unpublished data set in which gene expression is measured a few minutes after induction of each TF. We use one data set for learning a quantitative regulatory network and the other for testing TFA inference using that network. Two tests involve guessing which TF was perturbed in each expression profile, and whether the TF was activated or repressed. The third is to identify known examples of proteins regulating the activities of TFs. The fourth examines correlation between a TF’s inferred activity and its observed mRNA. Further analysis using a new, unpublished data set of TF binding obtained with the transposon calling cards method also found that the inferred strength with which a TF regulates each of its targets correlates positively with its measured binding strength at each target. With these data sets and metrics, we are able to determine, with high confidence, what works and what doesn’t in TFA inference. We also provide a validated, quantitative regulatory network for yeast.